| Легочный альвеолярный протеиноз | |
|---|---|
| Другие названия | Легочный альвеолярный протеиноз |
 |
|
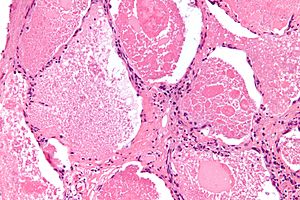
| Микрофотография легочного альвеолярного протеиноза, показывая характерное заполнение воздушного пространства очагово плотными глобулами, называемымиболтунамиилиплотными телами. пятно H&E | |
| Специальности | Пневмология |
Легочный альвеолярный протеиноз ( PAP ) — это редкое заболевание легких, характеризующееся аномальным накоплением в альвеолах легких липопротеиновых соединений, содержащих сурфактант. Накопленные вещества препятствуют нормальному газообмену и расширению легких, что в конечном итоге приводит к затруднению дыхания и восприимчивости к развитию легочных инфекций. Причины ЛПВП можно разделить на первичные (аутоиммунный ЛПВП, наследственный ЛПВП), вторичные (несколько заболеваний) и врожденные (несколько заболеваний, обычно генетических), хотя наиболее распространенной причиной является первичное аутоиммунное заболевание у человека.
Содержание
- 1 Признаки и симптомы
- 2 Причины
- 3 Генетика
- 4 Диагноз
- 5 Лечение
- 6 Эпидемиология
- 7 История
- 8 Исследование
- 9 Ссылки
- 10 Внешние ссылки
Признаки и симптомы
Признаки и симптомы ЛПВП включают одышку, [1] кашель, лихорадку низкого уровня и потерю веса Клиническое течение ЛПВП непредсказуемо. Спонтанная ремиссия признается, и у некоторых пациентов симптомы остаются неизменными. Смерть может наступить в результате прогрессирования ЛПВП или любого сопутствующего основного заболевания. Люди с ЛПВП более уязвимы к легочным инфекциям, таким как бактериальная пневмония, микобактериальная инфекция avium-intracellularis или грибковая инфекция. [цитата необходима]
Причины
Аномальное накопление липопротеиновых соединений в ЛПВП связано с нарушением регуляции и клиренса сурфактанта. Обычно это связано с нарушением функции альвеолярных макрофагов. [2] У взрослых наиболее распространенной причиной ПАП является аутоиммунитет к гранулоцитарно-макрофагальному колониестимулирующему фактору (ГМ-КСФ), критическому фактору развития альвеолярных макрофагов. Снижение биодоступности GM-CSF приводит к плохому развитию и функционированию альвеолярных макрофагов, что приводит к накоплению сурфактанта и сопутствующих продуктов. [2]
Вторичные причины ЛПВП — это те, при которых накопление липопротеиновых соединений является вторичным по отношению к другому процессу заболевания. Это было признано в условиях некоторых видов рака (например, миелоидной лейкемии), легочных инфекций или воздействия пыли или химических веществ, таких как никель. [3]
Хотя причина PAP изначально не была понятна, важным шагом вперед в понимании причины заболевания стало случайное наблюдение того, что у мышей, выведенных для экспериментальных исследований и лишенных гематологического фактора роста, известного как гранулоцит-макрофагальный колониестимулирующий фактор (GM-CSF), развился легочный синдром аномального накопления сурфактанта, похожий на человеческий PAP. [4]
Последствия этого открытия все еще изучаются, но в феврале 2007 года было сообщено о значительном прогрессе. Исследователи в этом докладе рассказали о наличии аутоантител к ГМ-КСФ у пациентов с PAP и продублировали этот синдром путем введения этих аутоантител мышам. [5]
Семейные или спорадические инактивирующие мутации в одном из родительских геновGATA2приводят к аутосомно-доминантному расстройству, называемому дефицитом GATA2. ГенGATA2производит транскрипционный фактор GATA2, который имеет решающее значение для эмбрионального развития, поддержания и функциональности кроветворения, лимфообразования и других тканеобразующих клеток. У людей с единственной инактивирующей мутациейGATA2наблюдается широкий спектр заболеваний, включая легочный альвеолярный протеиноз. GATA2Легочный альвеолярный протеиноз, обусловленный мутациями, связан с нормальным уровнем GM-CSF и обычно улучшается или предотвращается у больных людей, которые получают успешную трансплантацию гемопоэтических стволовых клеток. [6] [7]
Генетика
Наследственный легочный альвеолярный протеиноз (ЛПА) — это рецессивное генетическое заболевание, при котором люди рождаются с генетическими мутациями, нарушающими функцию рецептора CSF2 альфа на альвеолярных макрофагах. В результате молекула-мессенджер, известная как гранулоцит-макрофагальный колониестимулирующий фактор (GM-CSF), не может стимулировать альвеолярные макрофаги к выведению сурфактанта, что приводит к затруднению дыхания. Ген рецептора CSF2 альфа расположен в области 5q31 хромосомы 5, а продукт гена также может называться рецептором гранулоцитарно-макрофагального колониестимулирующего фактора. [8] [9]
Диагноз
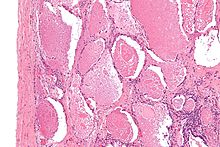
Диагноз PAP ставится на основании сочетания симптомов, визуализации грудной клетки и микроскопической оценки лаважа/легочной ткани. Для подтверждения полезно дополнительное тестирование на антитела к ГМ-КСФ в сыворотке крови. [10]
Хотя и симптомы, и результаты визуализации стереотипны и хорошо описаны, они неспецифичны и неотличимы от многих других заболеваний. Например, рентгенография грудной клетки может показать альвеолярные помутнения, а компьютерная томография — безумный рисунок легочной мостовой, причем оба этих признака чаще встречаются при множестве других заболеваний. [11] Поэтому диагноз зависит в первую очередь от результатов патологии. [цитата необходима]
Промывание легких или ткани для гистопатологического анализа чаще всего получают с помощью бронхоальвеолярного лаважа и/или биопсии легкого. [12] Характерные результаты биопсии показывают заполнение альвеол (и иногда терминальных бронхиол) аморфным эозинофильным материалом, который сильно окрашивается PAS-пятном и окрашиванием PAS-диастазы. Окружающие альвеолы и легочный интерстиций остаются относительно нормальными. [13] Электронная микроскопия образца, хотя она обычно не проводится из-за непрактичности, показывает пластинчатые тела, представляющие ПАВ. [14] Альтернативным диагнозом с аналогичными гистоморфологическими данными является пневмонияPneumocystis jirovicii. [14]
При промывании легких обычно выделяется жидкость, которая имеет «молочный» состав. Микроскопически в образцах видны PAS-положительные глобулы размером 20-50 микрометров на фоне мелкозернистого или аморфного PAS-положительного материала. Обычно наблюдается низкое количество макрофагов и воспалительных клеток (хотя этот показатель может варьироваться). [13] [14]
Лечение
Стандартным лечением ЛПВП является промывание всего легкого [15] [16] [17] и поддерживающая терапия. [18] [19] [20] Промывание всего легкого — это процедура, выполняемая под общей анестезией, при которой одно легкое накачивается кислородом (вентилируемое легкое), а другое легкое (невентилируемое легкое) заполняется теплым солевым раствором (до 20 л) и дренируется, удаляя вместе с ним любые белковые выделения. [21] Это обычно эффективно для улучшения симптомов ЛПВП, часто на длительный период времени. Другие методы лечения, которые еще находятся на стадии исследования, включают подкожное и ингаляционное введение ГМ-КСФ и ритуксимаб, внутривенное вливание, которое останавливает выработку аутоантител, ответственных за аутоиммунный ЛПВП. [18] [19] [20] [22] Трансплантация легких была проведена у людей с различными формами ЛПВП; однако часто к ней прибегают только тогда, когда все другие варианты лечения оказались безуспешными и развилось значительное повреждение легких из-за риска, осложнений или рецидива ЛПВП после трансплантации. [20] [22] [23]
Эпидемиология
Заболевание чаще встречается у мужчин и курильщиков табака. [цитата необходима]
В недавнем эпидемиологическом исследовании, проведенном в Японии [24], аутоиммунная ЛПВП имеет более высокую частоту и распространенность, чем сообщалось ранее, и не имеет тесной связи с курением, профессиональным воздействием или другими заболеваниями. липоидная пневмония и неспецифическая интерстициальная пневмония наблюдались до развития ЛПВП у ребенка. [25]
История
Впервые PAP был описан в 1958 году [26] врачами Самуэлем Розеном, Бенджамином Каслманом и Эвериллом Либоу. [27] В своей серии случаев, опубликованной в New England Journal of Medicine7 июня того же года, они описали 27 пациентов с патологическими доказательствами положительного материала Шиффа с периодической кислотой, заполняющей альвеолы. Этот богатый липидами материал позже был признан поверхностно-активным веществом. [цитата необходима]
Сообщение о лечении ЛПВП с помощью терапевтического бронхоальвеолярного лаважа было сделано в 1960 году доктором Хосе Рамиресом-Риверой в госпитале администрации ветеранов в Балтиморе [28], который описал многократное «сегментарное затопление» как средство физического удаления скопившегося альвеолярного материала. [29]
Поиск
PAP является одним из редких заболеваний легких, изучаемых в настоящее время Консорциумом по редким заболеваниям легких (RLDC). RLDC является частью Сети клинических исследований редких заболеваний (RDCRN), инициативы Управления исследований редких заболеваний (ORDR) Национального центра развития трансляционных наук (NCATS). RLDC занимается разработкой новых диагностических и терапевтических средств для пациентов с редкими легочными заболеваниями в сотрудничестве между Национальным институтом здравоохранения, организациями пациентов и клиническими исследователями. [цитата необходима]